Institute of Chemical Research of Catalonia (ICIQ), Tarragona, Catalonia, Spain
Computational chemistry has focused historically in the struggle for the accurate calculation of free energy profiles. Continued progress in computer power and theoretical methods has led in recent years to a situation where these free energy profiles have become rather accurate, in particular in domains such as computational homogeneous catalysis [1]. Because of this, we can now focus on further refinements to bring calculation closer to experiment. One of these refinements is microkinetic modeling, which allows the introduction of the effect of concentrations [2,3].
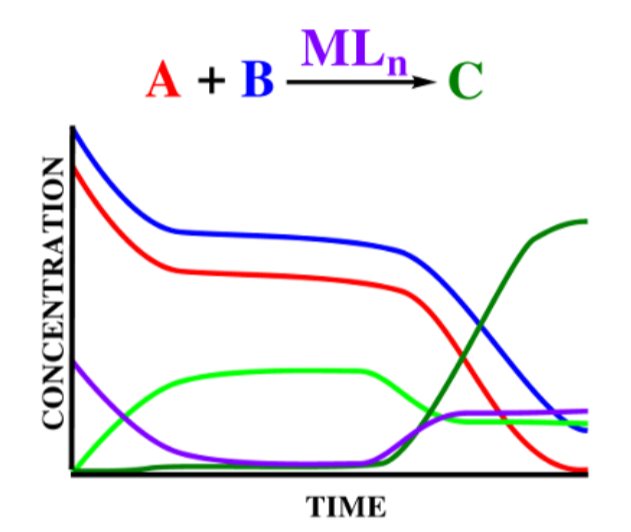
The raw experimental results usually involve reaction times rather than the energy barriers emerging from the free energy profiles. Microkinetic models can make the connection between rate constants calculated from density functional theory (DFT) and reaction times.
In this presentation we will briefly discuss the idea of the treatment, and present selected examples of application with COPASI, [4] one of the main codes freely available. Other extensions to free energy profiles, such as Marcus theory for single electron transfer steps will be be briefly discussed also.
Keywords: Density Functional Theory, Microkinetic modeling, Homogeneous catalysis.
[1] Harvey, J. N.; Himo, F.; Maseras, F.; Perrin, L. ACS Catal. 2019, 9, 6803-6813.
[2] Besora, M.; Maseras, F. WIREs Comput. Mol. Sci. 2018, 8, e1372.
[3] Sciortino, G.; Maseras, F. Top Catal. 2022, 65, 105-117.
[4] https://copasi.org (accessed Dec 9 th , 2023).
Removed per the speaker's request
The 2026 edition of the Virtual Winter School on Computational Chemistry will be proudly sponsored by:

The 2025 edition of the Virtual Winter School on Computational Chemistry was proudly sponsored by the School of Chemistry at the University of Edinburgh.
![]()
